The Terrier group study the mechanisms driving inflammation in autoinflammatory diseases and systemic vasculitis, especially large vessel vasculitis and ANCA-associated vasculitis.
Work on autoinflammatory diseases has focused primarily on VEXAS syndrome. Acquired mutations in the UBA1 gene, which occur in myeloid cells and result in the expression of a catalytically impaired isoform of the enzyme E1, have recently been identified in patients with a severe adult-onset autoinflammatory syndrome called VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic). The exact physiological and clinical effects of these mutations remain poorly defined. Using cell lines and patient cells, we are studying perturbations in inflammatory and cell death pathways induced by this mutation.
We aim to identify inflammatory pathways that are involved in VEXAS syndrome that could be targeted to reduce or prevent inflammation.
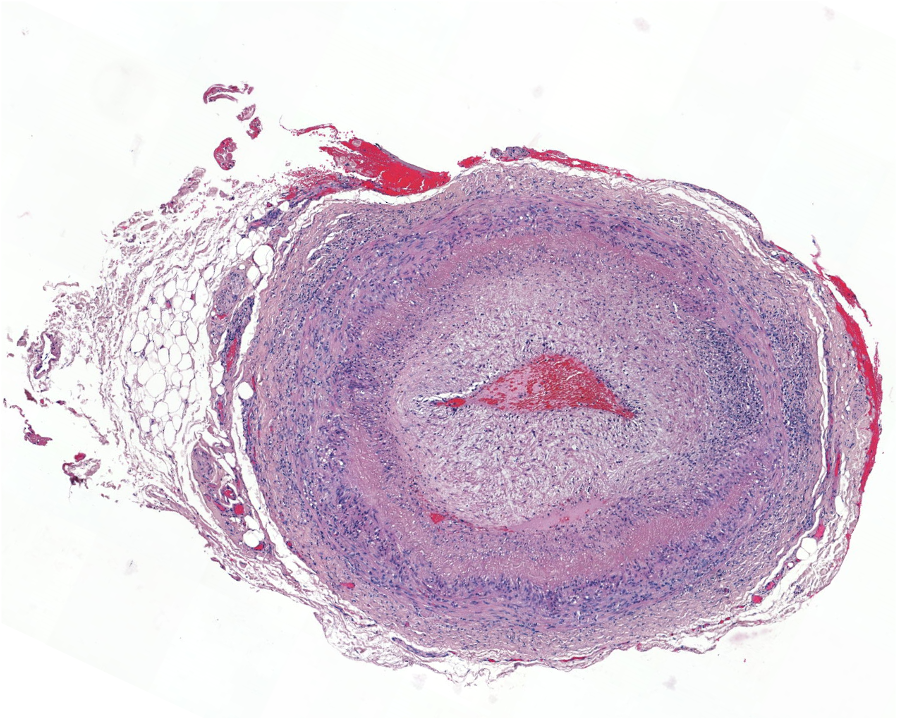
Work in systemic vasculitis focuses on large vessel vasculitis, primarily giant cell arteritis, the most common cause of vasculitis in adults over 50 years of age, and small vessel vasculitis, characterized by the presence of antineutrophil cytoplasmic antibodies (ANCA) and frequent involvement of the lungs and kidneys, called ANCA-associated vasculitis.
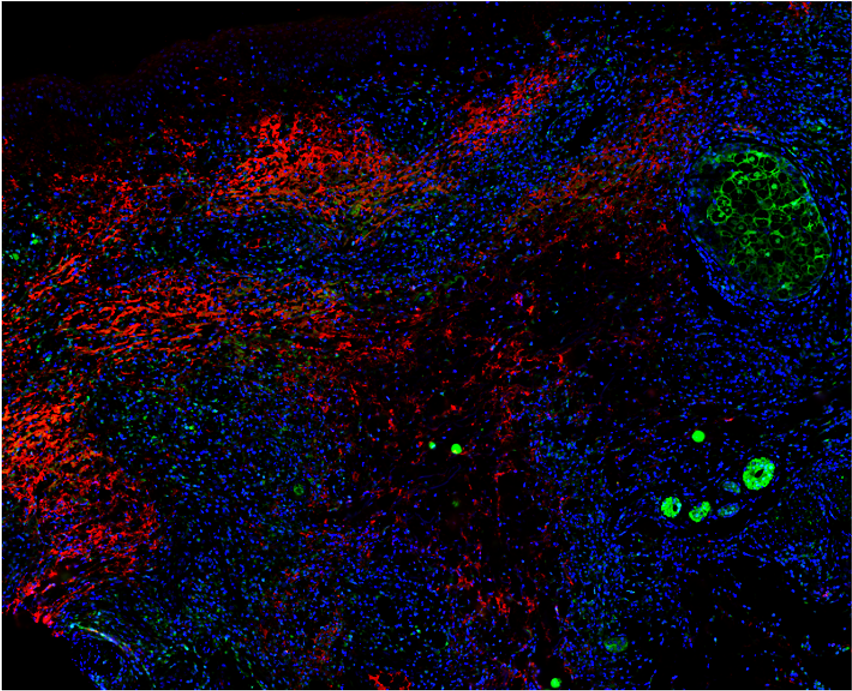
In large vessel vasculitis, we study the crosstalk between inflammatory cells and resident cells with the vessel wall. In ANCA-associated vasculitis, we are studying the inflammatory determinants of renal involvement, which is characterized by rapidly progressive glomerulonephritis, a severe form of kidney disease responsible for irreversible renal failure.
As previously, we aim to identify pathways that could be targeted to reduce or prevent inflammation and vascular damage in such diseases.
Recent Achievements
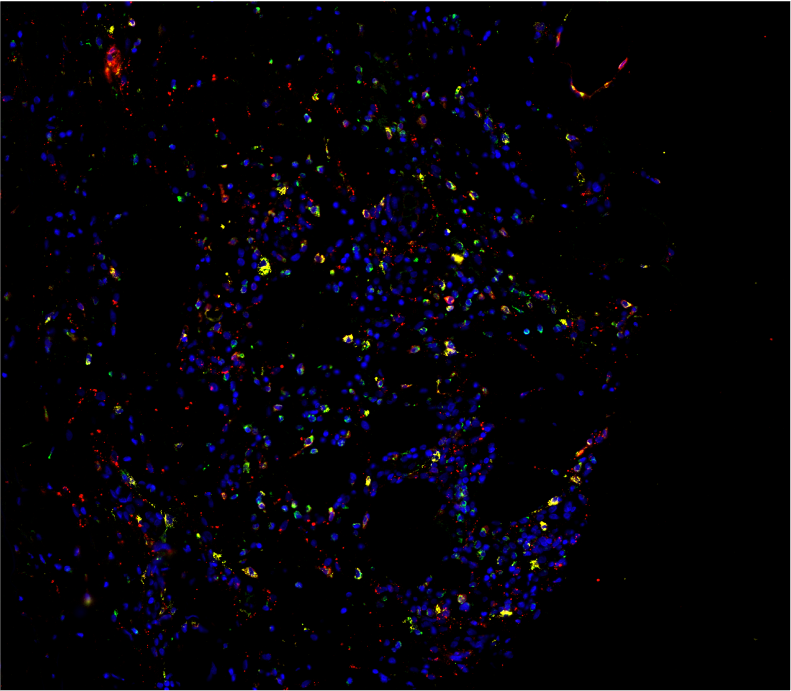
We dissected the mechanisms driving systemic inflammation in UBA1-mutant monocytes in VEXAS syndrome. We performed an integrated immune analysis including multiparameter phenotyping of peripheral blood leukocytes, cytokine profiling, bulk and single cell gene expression analysis, and skin tissue imaging mass cytometry. We showed that monocytes from UBA1 mutant individuals were quantitatively and qualitatively impaired and exhibited features of exhaustion with aberrant expression of chemokine receptors. In the peripheral blood of VEXAS patients, we identified a significant increase in circulating levels of many proinflammatory cytokines, including IL-1β and IL-18, which reflect inflammasome activation and markers of myeloid cell dysregulation. Whole blood gene expression analysis confirmed the role of circulating cells in IL-1β and IL-18 dysregulation in VEXAS patients and revealed a significant enrichment of TNF-a and NFkB pathways that could mediate cell death and inflammation. Single-cell analysis confirmed the inflammatory state of monocytes from VEXAS patients and allowed us to identify specific molecular pathways that could explain monocytopenia. Taken together, these findings provide important insights into the molecular mechanisms involved in mature myeloid commitment in VEXAS syndrome and suggest that control of the undescribed inflammasome activation and inflammatory cell death may be novel therapeutic targets in this disease.
Kosmider et al. VEXAS syndrome is characterized by inflammasome pathway activation and monocyte dysregulation. Nature Communications. In press.