TEAM LEADER : Pierre-Louis Tharaux & Olivia Lenoir
Mail : pierre-louis.tharaux@inserm.fr or olivia.lenoir@inserm.fr
Olivia LENOIR: +33 1 53 98 80 19 / +33 1 53 98 80 19
Localisation : 2nd floor, room 219 & 220
DOCTORAL SCHOOL : Ecole Doctorale Bio Sorbonne Paris Cité (BioSPC)


The Lenoir group study the pathophysiological mechanisms of kidney and vessels injury in common chronic kidney diseases (CKD). Despite the strong prevalence of diabetic kidney disease worldwide and the emergence of the recent sodium glucose cotransporter 2 inhibitors and glucagon-like peptide 1 receptor agonists, providing significant but only partial efficacy to prevent cardiovascular events and kidney failure, there are currently no therapies to specifically prevent glomerular cells injury during diabetes and hypertension.
We aim to identify mechanisms of kidney and vessels tolerance during diabetes and hypertension.
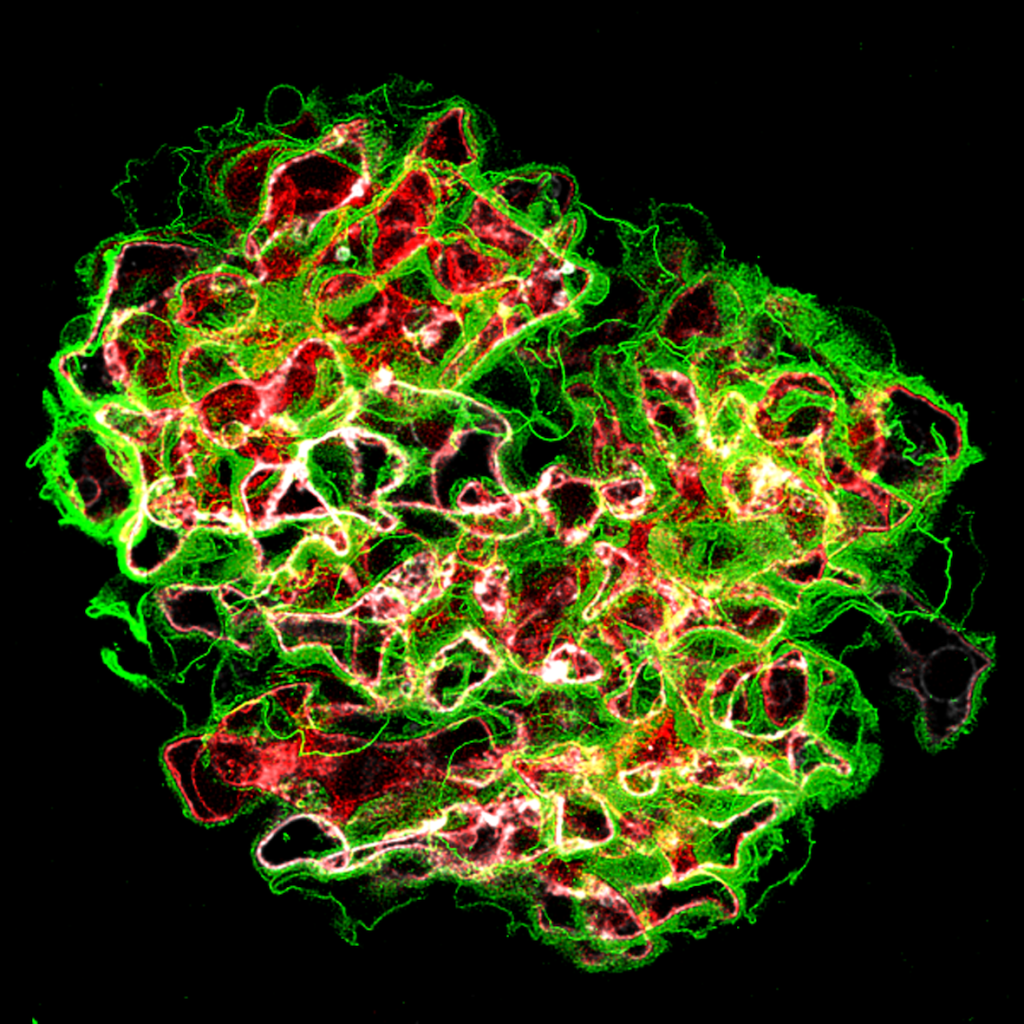
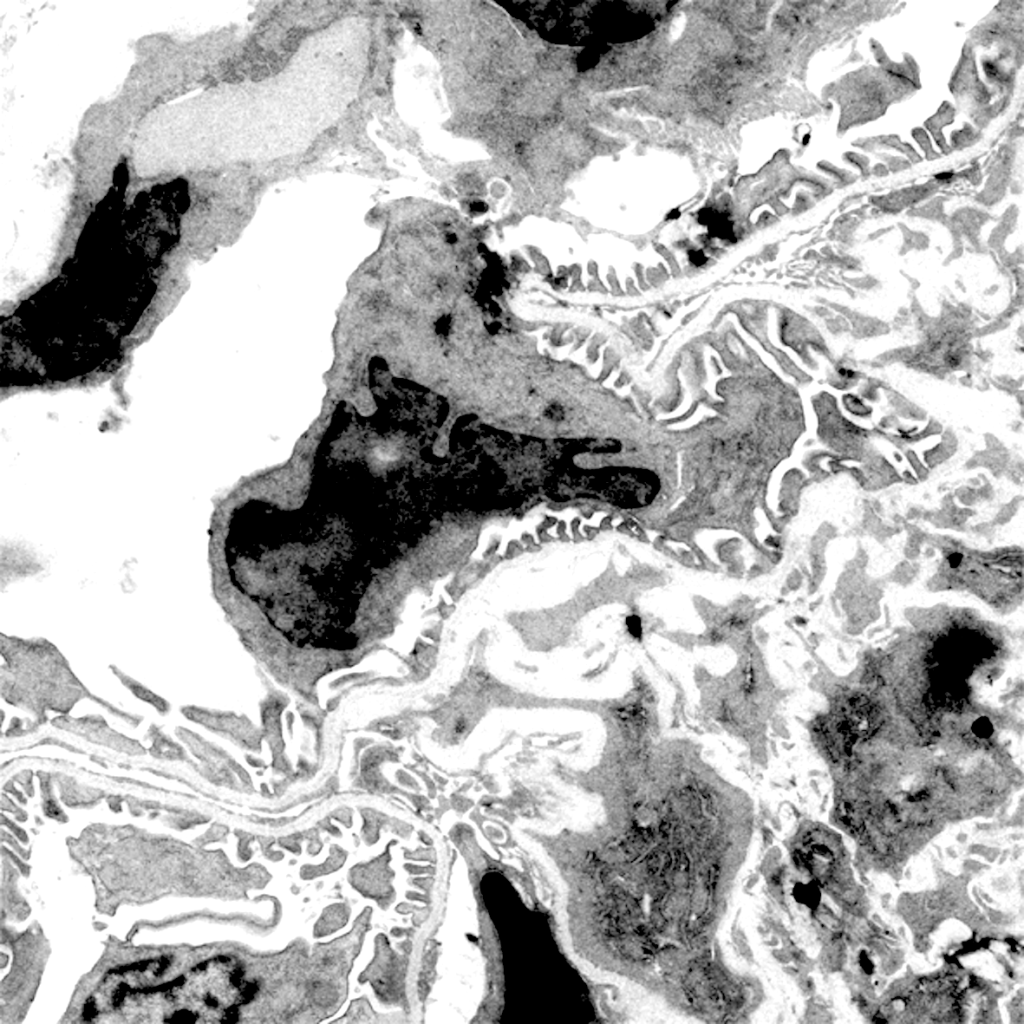
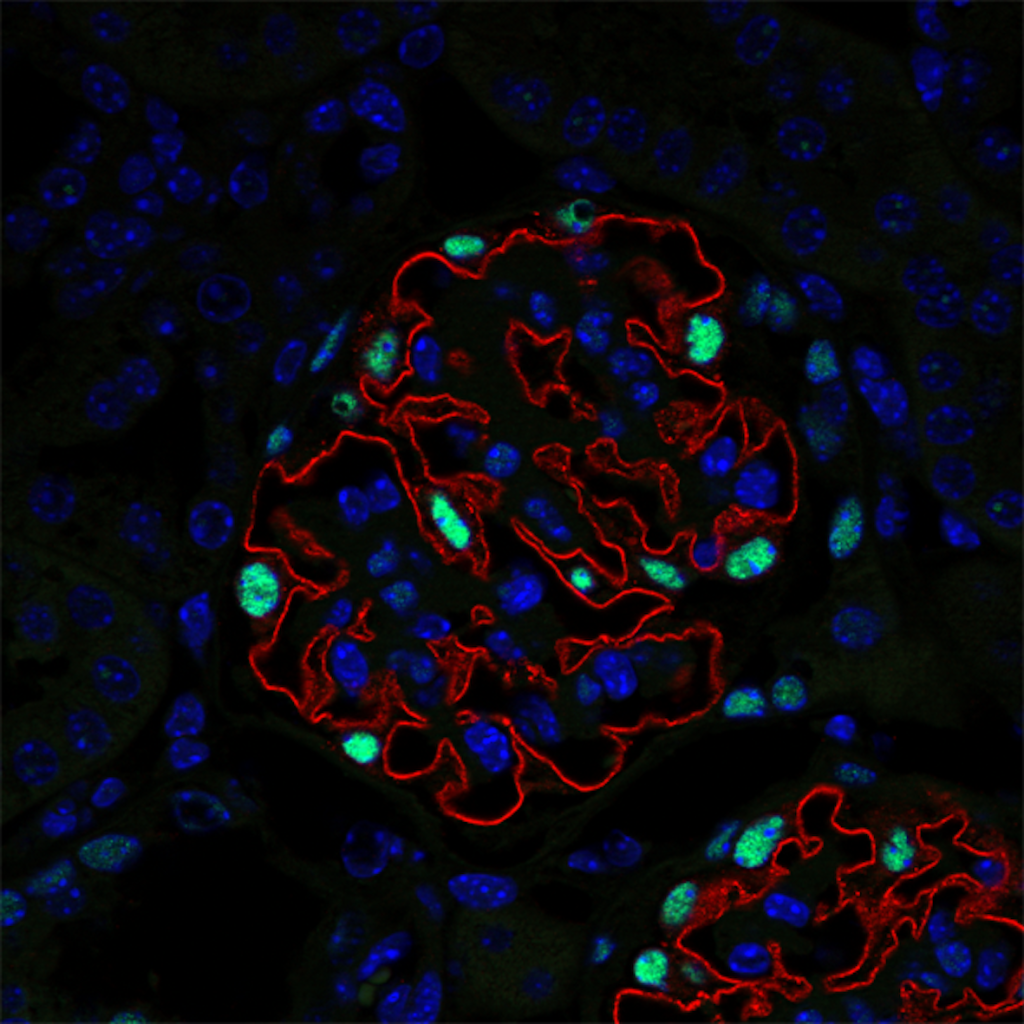
Developing new tools to study cell-cell interactions within glomeruli is a necessity. Animal models are informative, but they are increasingly criticized by the civil society. In addition, they do not allow to monitor phenotypic changes in cells in real time. In collaboration with the Ecole Polytechnique (LOB Laboratory, C. Bouzigues), we are developing a glomerulus-on-a-chip, enabling to study phenotypic changes in glomerular cells and the function of the glomerular filtration barrier during the mimicked pathology. We also develop Imaging Mass Cytometry and image analysis algorithms to study the multiple phenotypic switches and cellular cross-communications in kidneys in human pathologies.
Autophagy is a highly regulated lysosomal protein degradation pathway that removes protein aggregates and damaged or excess organelles to maintain intracellular homeostasis and cell integrity. Growing evidence suggests that autophagy is implicated in cell injury during renal diseases, both in the tubulointerstitial compartment and in glomeruli. We previously demonstrated that the autophagy gene, Atg5, is essential in endothelial cells and podocyte to maintain kidney microvascular integrity during diabetes mellitus in mice (Lenoir et al. Autophagy 2015). In collaboration with T. Huber’s group we further demonstrated that unlike the majority of cells whose autophagy is regulated by the mTOR pathway, autophagy in podocytes is regulated independently of the mTOR pathway (Bork et al. Autophagy 2020).
We aim to understand the mechanisms regulating autophagy in glomerular cells and the pharmacological means of modulating autophagy in CKD.
We highlighted mechanisms of podocyte injury in chronic kidney disease linking the hypertensive and diabetic environment to the dysregulation of podocyte autophagy. We demonstrated that aberrant TRPC6-mediated calpain activation in podocytes promotes autophagy impairment and subsequent podocyte injury. Calpain or TRPC6 inhibition restored podocyte autophagy, limiting podocyte injury in disease models of hypertension and diabetes.
We unraveled a fundamental role of autophagy in endothelial function, linking cell proteostasis to mechanosensing. We demonstrated that autophagy regulates VEGF-dependent VEGFR signaling and VEGFR-mediated and flow-mediated eNOS activation. Endothelial ATG5 deficiency in vivo resulted in selective loss of flow-induced vasodilation in mesenteric arteries and kidneys and increased cerebral and renal vascular resistance in vivo. We found a crucial pathophysiological role for autophagy in endothelial cells in flow-mediated outward arterial remodeling, prevention of neointima formation following wire injury, and recovery after myocardial infarction.